
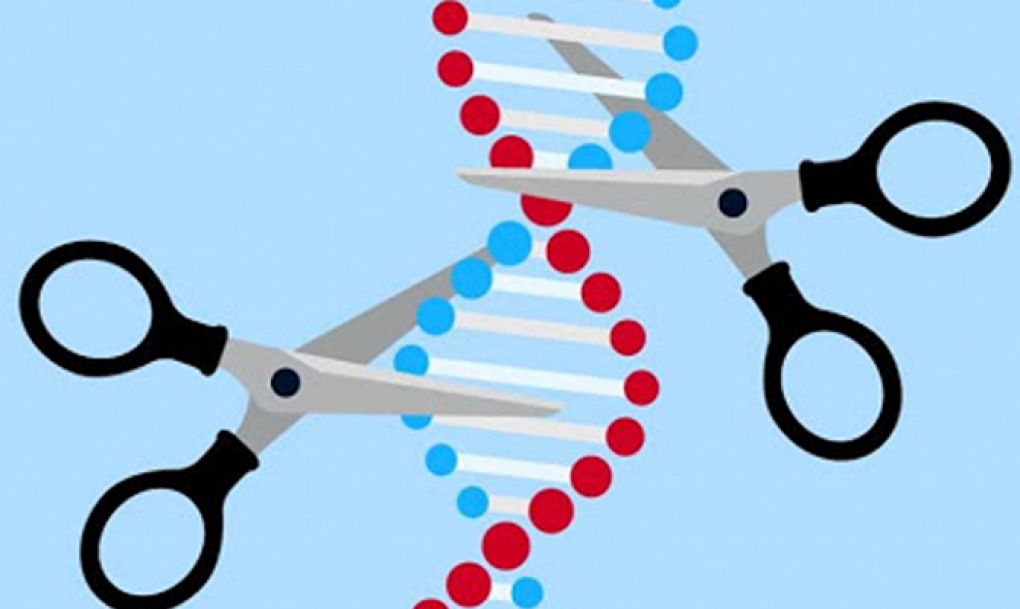
La Agencia Europea del Medicamento (EMA, por sus siglas en inglés) ha recomendado este viernes la aprobación del primer medicamento que utiliza la novedosa tecnología de edición de genes CRISPR/Cas9.
La terapia, de nombre comercial Casgevy, está indicada para el tratamiento de la beta talasemia dependiente de transfusiones y la anemia falciforme grave en pacientes de 12 años o más para quienes es apropiado un trasplante de células madre hematopoyéticas, pero no existe un donante adecuado disponible. Según se explica desde la EMA, este nuevo medicamento basado en la tecnología CRISPR tiene el potencial de mejorar significativamente su calidad de vida. En este sentido, puede liberar a los afectados de la carga de las transfusiones frecuentes y las dolorosas crisis vasooclusivas (CVO) que ocurren cuando los glóbulos rojos falciformes bloquean los vasos sanguíneos pequeños.
La beta talasemia y la anemia de células falciformes (SCD, por sus siglas en inglés) son dos enfermedades raras hereditarias causadas por mutaciones genéticas que afectan la producción o función de la hemoglobina, la proteína que se encuentra en los glóbulos rojos y que transporta oxígeno por todo el cuerpo. Ambas patologías son debilitantes y potencialmente mortales durante toda la vida.
Tecnología CRISPR para editar células madre del propio paciente
Casgevy es una terapia génica basada en células medicamento que utiliza tecnología CRISPR/Cas9 para editar las células madre sanguíneas del propio paciente. Se trata de un tratamiento único y personalizado que consiste en movilizar células madre de la médula ósea a partir de la sangre de la persona.
La edición de genes CRISPR encuentra una secuencia específica de ADN dentro de una célula. Utilizando tijeras moleculares para realizar cortes precisos, permite agregar, eliminar o alterar material genético en esa ubicación específica del genoma de las células.
Con Casgevy, las células madre se editan en la región potenciadora específica de los eritroides del gen BCL11A, que normalmente previene la producción de hemoglobina fetal (HbF). Posteriormente, estas células modificadas se infunden nuevamente en el paciente, y la reducción de la transcripción del gen BCL11A conduce a un aumento de la producción de HbF, proporcionando así hemoglobina funcional.
Casgevy recibió apoyo a través del esquema PRIority MEdicines (PRIME) de la EMA, que proporciona apoyo científico y normativo temprano y reforzado a los medicamentos que tienen un potencial especial para satisfacer las necesidades médicas no cubiertas de los pacientes.
Recomendación basada en dos ensayos clínicos
La Agencia Europea ha basado su recomendación en dos ensayos en curso de un solo grupo en pacientes de 12 a 35 años. En el primero, se incluyeron en el conjunto primario de eficacia 42 pacientes —incluidos 13 adolescentes— con beta talasemia dependiente de transfusiones, que recibieron una dosis única. De estos 42 pacientes, 39 estuvieron libres de transfusiones durante al menos un año.
En el segundo ensayo, se incluyeron en el conjunto primario de eficacia 29 pacientes que padecían anemia de células falciformes grave, seis de ellos adolescentes. De estos 29 afectados, 28 estuvieron libres de episodios de crisis vasooclusivas (CVO) durante al menos 12 meses consecutivos. Caracterizadas por causar dolor intenso y daño orgánico, las CVO son la principal causa de visitas a Urgencias y de hospitalizaciones en pacientes con esta enfermedad (SCD).
En cuanto a la seguridad de la nueva terapia, la EMA explica que se evaluó en los mismos dos ensayos de un solo brazo en curso y en un estudio de seguimiento a largo plazo, en los que 97 pacientes adolescentes y adultos con beta talasemia dependiente de transfusión o SCD fueron tratados con el medicamento.
Autorización de comercialización condicional
Los efectos secundarios más comunes asociados a este tratamiento basado en CRISPR son recuentos bajos de glóbulos blancos —incluida neutropenia febril—, niveles bajos de plaquetas, enfermedad hepática, náuseas, vómitos, dolor de cabeza y llagas en la boca. Estos eventos se deben a los medicamentos necesarios para que las células sanguíneas modificadas se injerten y reemplacen a las células madre no modificadas.
En este escenario, la Agencia Europea del Medicamento recomienda Casgevy para una autorización de comercialización condicional, uno de los mecanismos regulatorios de la Unión Europea para facilitar el acceso temprano a medicamentos que satisfacen una necesidad médica no cubierta.
Este tipo de aprobación permite a la EMA recomendar una terapia para autorización con datos menos completos de lo esperado normalmente, si el beneficio de su disponibilidad inmediata para los pacientes supera el riesgo inherente al hecho de que no todos los datos estén todavía disponibles.
Para confirmar la eficacia y la seguridad de Casgevy, desarrollada por la empresa suiza CRISPR Therapeutics, la compañía tendrá que presentar antes de agosto de 2026 los resultados finales obtenidos en los ensayos pivotales actualmente en curso, así como los resultados del estudio de seguimiento a largo plazo que se está desarrollando y otros análisis del producto que se llevarán a cabo.
La Comisión Europea decidirá sobre la recomendación
Los pacientes tratados con Casgevy se someterán a seguimiento durante 15 años para controlar la eficacia y seguridad a largo plazo de esta terapia génica. La empresa también tendrá que realizar y presentar los resultados de un estudio basado en los datos de un registro de pacientes.
En su evaluación global de los datos disponibles, el Comité de Terapias Avanzadas (CAT), grupo de expertos de la EMA para medicamentos celulares y genéticos, ha considerado que los beneficios de este nuevo tratamiento superaban los posibles riesgos en pacientes con beta talasemia y anemia falciforme. Por su parte, el Comité de Medicamentos de Uso Humano de la Agencia Europea (CHMP) se ha mostrado de acuerdo con la evaluación y el dictamen positivo del CAT, por lo que ha recomendado su aprobación.
El dictamen del CHMP es un paso intermedio en el camino de Casgevy hacia el acceso de los pacientes. El informe se enviará ahora a la Comisión Europea para que adopte una decisión sobre la autorización de comercialización en toda la UE. Una vez concedida, las decisiones sobre el precio del medicamento y el reembolso corresponderán a cada Estado miembro, teniendo en cuenta el papel o el uso potencial de esta terapia en el contexto del sistema sanitario nacional de ese país.













Recordamos que SALUD A DIARIO es un medio de comunicación que difunde información de carácter general relacionada con distintos ámbitos sociosanitarios, por lo que NO RESPONDEMOS a consultas concretas sobre casos médicos o asistenciales particulares. Las noticias que publicamos no sustituyen a la información, el diagnóstico y/o tratamiento o a las recomendaciones QUE DEBE FACILITAR UN PROFESIONAL SANITARIO ante una situación asistencial determinada.
SALUD A DIARIO se reserva el derecho de no publicar o de suprimir todos aquellos comentarios contrarios a las leyes españolas o que resulten injuriantes, así como los que vulneren el respeto a la dignidad de la persona o sean discriminatorios. No se publicarán datos de contacto privados ni serán aprobados comentarios que contengan 'spam', mensajes publicitarios o enlaces incluidos por el autor con intención comercial.
En cualquier caso, SALUD A DIARIO no se hace responsable de las opiniones vertidas por los usuarios a través de los canales de participación establecidos, y se reserva el derecho de eliminar sin previo aviso cualquier contenido generado en los espacios de participación que considere fuera de tema o inapropiados para su publicación.
* Campos obligatorios